3. Molecular Dynamics Simulation¶
MD Simulation¶
Molecular dynamics (MD) simulations were performed to study the time-dependent stability and binding behavior of peptide–HLA-B27 complexes. MD allows observation of molecular motions beyond static docking models and provides insight into conformational stability, flexibility, and interaction persistence under physiological conditions.
In this study, MD simulations were used to compare the dynamic behavior of one human peptide (anx) and three microbial peptides (kp1, kp2, kp3) when bound to HLA-B27.
System Preparation¶
Docked peptide–HLA-B27 complexes were prepared for MD simulations using AmberTools23 Case et al. (2023),. The docking output structures were cleaned, protonated, and standardized to ensure compatibility with classical force-field–based simulations.
A custom preparation workflow was applied to convert docking models into Amber-ready structures, followed by solvation and ion neutralization.
Force Field and Solvation¶
Protein parameters were assigned using the ff14SB force field. Each complex was solvated in a TIP3P water box with periodic boundary conditions. Counter ions (Na⁺ and Cl⁻) were added to neutralize the system and mimic physiological ionic strength.
Figure 1 shows the Solvated system of peptide–HLA-B27 complex before MD simulation
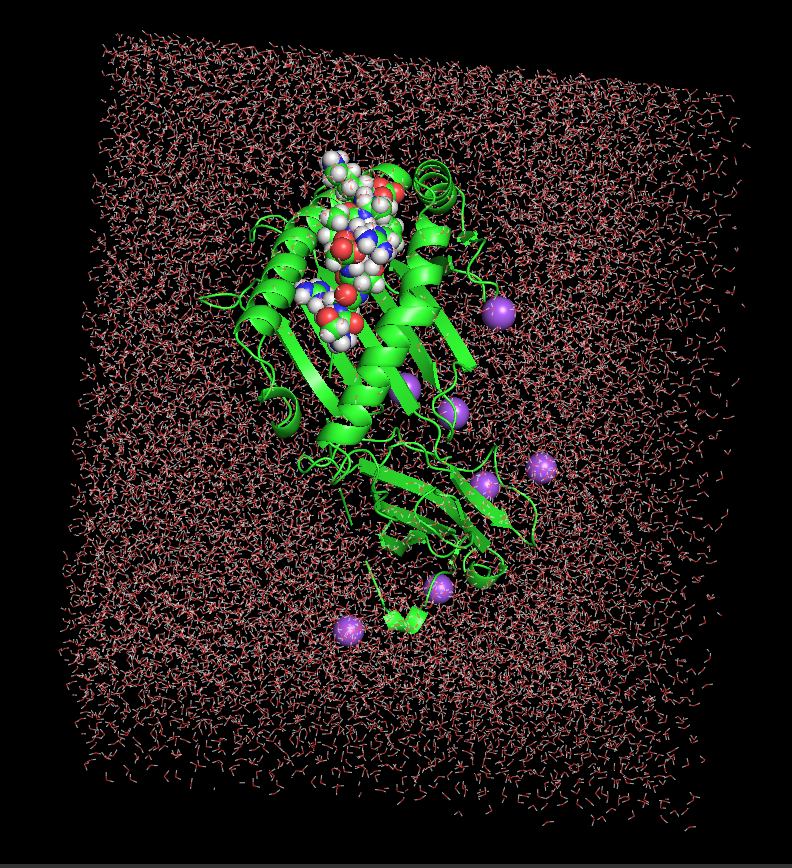
Figure 1:Solvated peptide–HLA-B27 system before MD simulation
MD Simulation Protocol¶
All MD simulations were performed using GROMACS 2024 Abraham et al. (2015),. After system preparation, each complex underwent a standard multi-step MD protocol consisting of energy minimization, equilibration, and production simulation.
Energy minimization was performed to remove steric clashes and unfavorable contacts. This was followed by equilibration under constant volume (NVT) and constant pressure (NPT) conditions to stabilize temperature and pressure. Finally, production MD simulations were carried out to sample biologically relevant conformational dynamics.
Simulation Conditions¶
Simulations were performed at a temperature of 300 K and a pressure of 1 bar. Long-range electrostatics were treated using the Particle Mesh Ewald (PME) method, and a time step of 2 fs was used. Production simulations were run for up to 1 μs for each complex.
- Case, D. A., Aktulga, H. M., Belfon, K., Ben-Shalom, I., & others. (2023). AmberTools. Journal of Chemical Information and Modeling. 10.1021/acs.jcim.3c00206
- Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., & others. (2015). GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX, 1–2, 19–25. 10.1016/j.softx.2015.06.001