2. Molecular Docking¶
Molecular docking was performed to model peptide–HLA-B27 complexes and evaluate the binding orientation of selected human and microbial peptides within the HLA groove.
Docking was carried out using AlphaFold-Multimer Jumper et al. (2021) and HADDOCK Dijk & Bonvin (2003), via web server, which was used to generate structural models of peptide–HLA-B27 complexes. For each selected Annexin (ANX) peptide, all shortlisted Klebsiella pneumoniae (KP) peptides were docked individually with HLA-B27.
Output and Model Evaluation¶
After docking the AlphaFold-Multimer generated:
Structural models in
.cifformat.Confidence scores in accompanying
.jsonfiles.
The results of docking process were evaluated based on:
Predicted docking confidence.
Structural plausibility of the complex.
Peptide orientation and positioning within the HLA-B27 binding groove
Selection of Peptides for MD Simulations¶
Based on docking confidence scores and visual inspection, three microbial peptides were selected against one human Annexin peptide.
The final peptide set consisted of four peptides:
Human peptide
IRSEFKRKY (anx)
Microbial peptides
GRSDFKGDY (KP1)
GRSDFKGDYF (KP2)
SRSDRVAKY (KP3)
Figure 1 shows the docked structural models of the four selected peptide–HLA-B27 complexes used for subsequent MD simulations.
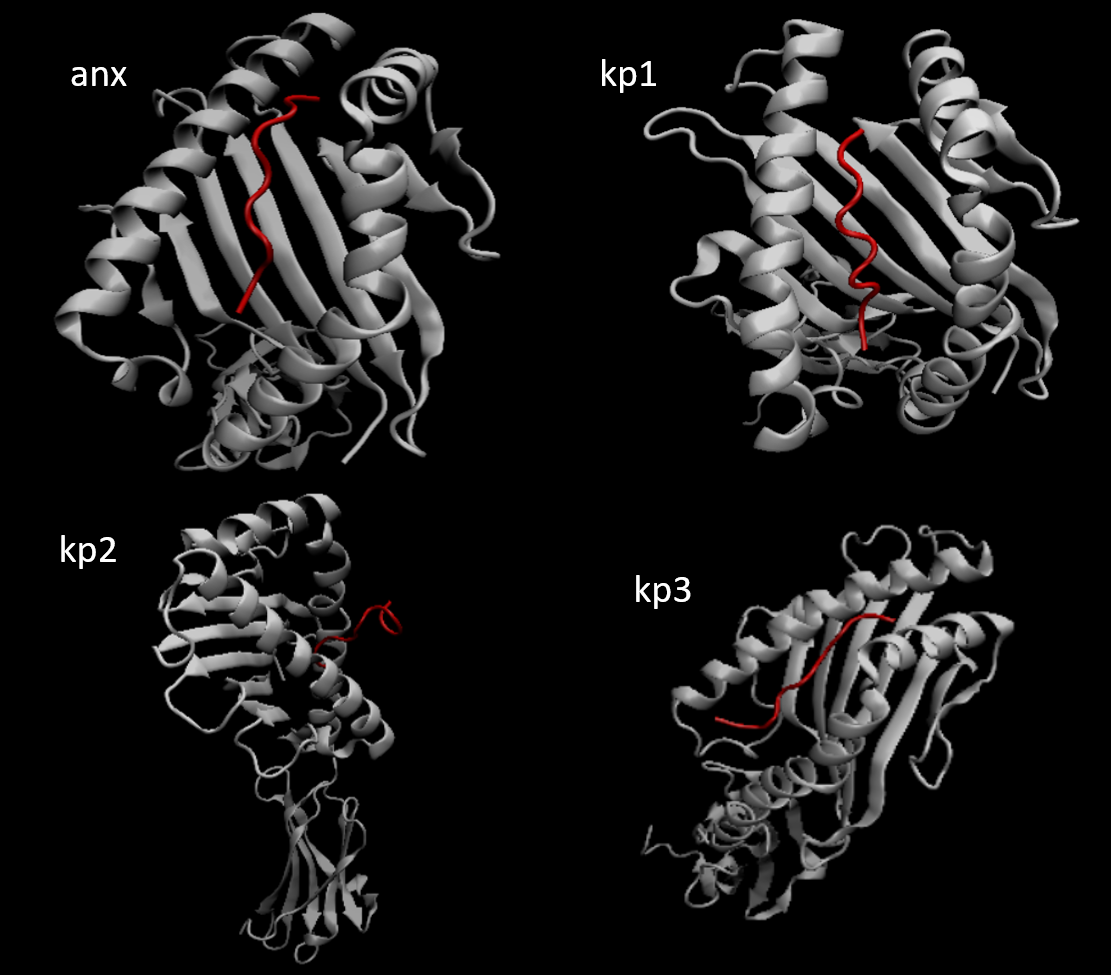
Figure 1:Model of peptides generated on PyMol
These peptides showed stable and well-positioned docking poses within the HLA groove and were selected for molecular dynamics simulations.
- Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., & others. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596, 583–589. 10.1038/s41586-021-03819-2
- van Dijk, M., & Bonvin, A. M. J. J. (2003). HADDOCK: a protein–protein docking approach based on biochemical or biophysical information. Journal of the American Chemical Society, 125(47), 14668–14669. 10.1021/ja0369397
- Schrödinger, LLC. (2022). The PyMOL Molecular Graphics System, Version 2.5.